
If you’re a medical device manufacturer or distributor looking to enter the Malaysian market, your product must go through medical device registration before it can be legally imported, exported, or sold in the country.
Malaysia’s medical device regulatory system is one of the most structured in Southeast Asia, and understanding it thoroughly can mean the difference between a smooth market entry and months of costly delays. This guide breaks down everything you need to know about MDA registration from device classification and required documents to fees and realistic approval timelines.
What Is MDA Registration in Malaysia?
Malaysia’s healthcare sector continues to grow rapidly, supported by a robust regulatory environment aimed at ensuring the safety, quality, and performance of medical devices. Central to this framework is the Medical Device Act 2012 (Act 737). Under this Act, all medical devices must be registered with the Medical Device Authority (MDA) before they can be imported, exported, or marketed in Malaysia.
The MDA is a federal statutory agency under the Ministry of Health (MoH) that implements and enforces the regulations as per the Medical Device Act 2012. In short, if you’re selling medical products in Malaysia whether locally manufactured or imported, MDA registration is a legal requirement.
Key Requirements Before MDA Registration
Before you even think about submitting an application, a few essentials need to be in place.
Appointing a Local Authorized Representative
This is a step that many foreign manufacturers overlook, but it’s actually the very first thing you need to do. Because foreign manufacturers cannot register directly with the MDA, the AR acts as the official liaison and the legal entity responsible for holding the device registration. The AR is responsible for registration, post-market surveillance, and communication with the MDA. Without an AR, you cannot begin the registration process.
The AR must be located in Malaysia and must have a valid AR license issued by the MDA. The AR must also have a sound understanding of Malaysian medical device regulations.
For distributors like Octopus Distribution, acting as or partnering with a qualified AR is one of the most valuable services you can offer to your overseas principal, it removes a major regulatory barrier and accelerates the path to market.
Getting Your Device Classification Right
You need to determine which risk class your device falls into. This single factor determines everything, the pathway you follow, the documents you need, and how long the process will take.
Medical device classification in Malaysia aligns with ASEAN device classification rules. Devices and IVDs are classified into Class A, B, C, and D by risk level. Getting the classification right is critical. Misclassifying a device even unintentionally can result in submission rejections and significant delays.
| Class | Risk Level | Examples |
| A | Lowest | Bandages, exam gloves, tongue depressors |
| B | Low-Moderate | Hypodermic needles, suction equipment |
| C | Moderate-High | Lung ventilators, bone fixation plates |
| D | Highest | Heart valves, implantable defibrillators |
Conformity Assessment for Higher-Risk Devices
If your product falls under Class B, C, or D, there’s an extra step which is evaluation by a Conformity Assessment Body (CAB).
Before your device reaches the Medical Device Authority, it must first pass through this third-party review. This independent review checks that your device meets Malaysia’s safety and performance standards. A CAB doesn’t just skim through your documents, it conducts a structured, in-depth assessment of your device and your quality systems.
This typically includes:
- Reviewing your technical documentation (CSDT)
- Verifying compliance with Essential Principles of Safety and Performance
- Auditing your Quality Management System (ISO 13485)
- Evaluating risk management and clinical data (for higher-risk devices)
- Checking labeling, instructions for use, and claims
For lower-risk devices (Class A), the regulatory burden is lighter. But as risk increases, so does scrutiny. For Class B, C, and especially Class D devices, regulators need more assurance because the impact on patient safety is higher, the device may be invasive or life-supporting and any failure could have serious consequences. That is why the CAB acts as a technical gatekeeper, ensuring only compliant products move forward.
Required Documents for MDA Registration
Document preparation is where most applications run into trouble. The requirements differ significantly between device classes.
For Class A Devices (Low Risk)
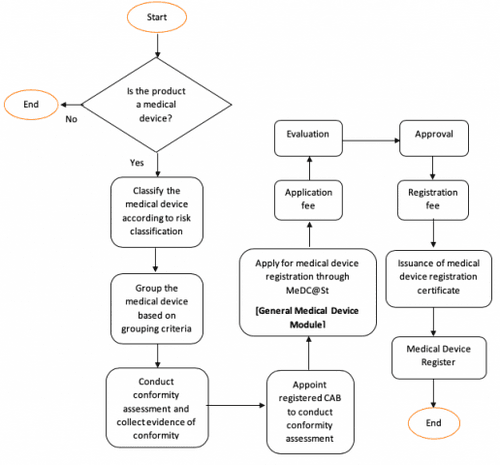
For Class A devices, which are deemed low-risk, a Conformity Assessment Body (CAB) evaluation is not required. Applicants submit documents directly to the MDA. The core documents required include:
- Manufacturer’s ISO Certificate – proof of a certified Quality Management System, typically ISO 13485
- Declaration of Conformity (MDA Format) – a formal declaration that the product complies with the Malaysian Medical Device Regulation 2012, formatted as per Appendix 1A
- Product Catalogue – marketing materials describing the product, specifications, intended use, and available variants
- Post-Market Vigilance History (last 3 years) – a report detailing any adverse events, recalls, or field safety corrective actions
- Letter of Authorization (LOA) – required if the signatory is not part of the top management
- LOA to Appoint Local Agent as AR – a formal letter designating the establishment applicant as the AR in Malaysia
- Pre-Market Approval (if applicable) – valid certification from recognised authorities such as the US FDA, CE (EU), TGA (Australia), Canada TPD, or Japan MHLW (not mandatory, but strengthens the application)
For Class B, C, and D Devices (Moderate to High Risk)
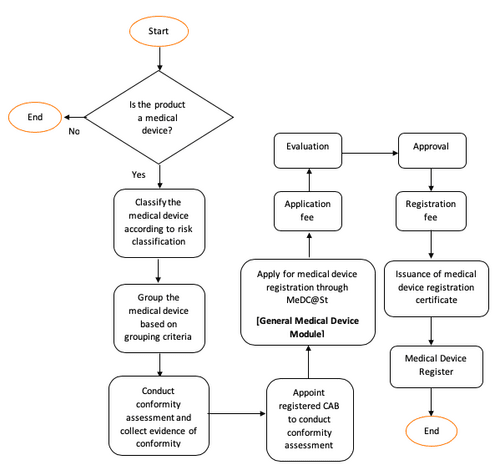
For Class B, C, and D medical devices in Malaysia, a Conformity Assessment Body (CAB) must first evaluate the technical documentation. Once the CAB issues a favourable assessment, the documents are then submitted to the Medical Device Authority (MDA) for final approval.
The documentation package for higher-risk classes is considerably more comprehensive:
- Manufacturer’s ISO 13485 Certificate – proof of an active and valid Quality Management System
- CSDT (Common Submission Dossier Template) – must follow the MDA template and guidance document precisely
- Essential Principles of Safety and Performance of Medical Device
- Declaration of Conformity (MDA Format)
- Pre-Market Clearance/Approval by Other Authorities (if applicable) – certificates from the FDA, CE, TGA, Canada TPD, or Japan MHLW
- Sterilization Validation Report / Calibration Certificate / IEC 60601 Test Certificate / Biocompatibility Report (where applicable)
- CAB Assessment Report and Certificate
General Labelling Requirements
The MDA registration number and contact information of the Malaysia AR must be included on product labels. Additionally, labelling for lay-use devices must be in Bahasa Melayu and may also include English.
MDA Registration Fees in Malaysia
MDA fees are split into application fees (when you submit) and registration fees (when you’re approved).
| Device Class | Application Fee (Non-refundable) | Registration Fee (Upon Approval) |
| Class A | RM 500 (Revised from RM 100) | RM 750 (New for 2026) |
| Class B | RM 250 | RM 1,000 |
| Class C | RM 500 | RM 2,000 |
| Class D | RM 750 | RM 3,000 |
Hidden Costs to Keep in Mind
Beyond official fees, there are additional costs that often catch companies off guard:
- CAB assessment fees
- Authorized Representative service charges
- Documentation preparation
MDA Registration Timeline
Timelines vary considerably depending on your device class and how well your dossier is prepared.
| Stage | Class A | Class B | Class C & D |
| Establishment License | 4–6 weeks | 4–6 weeks | 4–6 weeks |
| CAB Assessment | Not required | ~3–4 months | ~4–6 months |
| MDA Review | 2–3 months | 3–6 months | 6–12 months |
| Estimated Total | 3–4 months | 6–10 months | 10–18 months |
Several factors can influence your timeline:
- Completeness of your documentation
- How quickly your CAB completes its review
- Questions or clarifications from MDA
- Complexity of your device
Even a small inconsistency can add weeks (or months) to your approval. Once approved, your registration is valid for 5 years and renewal is required.
Frequently Asked Questions
Starting January 1, 2026, the MDA has significantly revised the fee structure for Class A devices. Previously, these were the most affordable to register, but the application fee has increased from RM 100 to RM 500, and a new approval-stage registration fee of RM 750 has been introduced. Fees for Class B, C, and D remain consistent with the tiered risk system.
Some of the most common issues include incorrect device classification, missing technical documents, poorly structured CSDT submissions or lack of local regulatory knowledge.
MDA registration ensures your device meets Malaysia’s safety standards, can legally enter the market and builds trust with healthcare providers.
No. Foreign manufacturers must appoint a Malaysia-based Authorized Representative (AR).The AR is responsible for submitting the application, communicating with MDA and ensuring ongoing regulatory compliance. Without an AR, your application cannot proceed.
Once approved, a medical device registration is typically valid for five years. You must initiate the renewal process via MeDC@St at least 6 to 9 months before the expiry date to avoid market interruption.
Planning to Register Your Medical Device in Malaysia?
If you’re entering the Malaysian market, it helps to have a partner who knows the process inside out. From classification and documentation to CAB coordination and final approval, every step plays a role in your success.
At Octopus Distribution, we support manufacturers in navigating the MDA process efficiently, minimizing delays, and ensuring compliance from start to finish. Get in touch today and we’ll help you map out a clear, practical path to registration so you can focus on growing your business.








